ZusammensetzungWirkstoffe
Apremilast.
Hilfsstoffe
Mikrokristalline Cellulose, Lactose-Monohydrat (60 mg / 120 mg / 180 mg pro Filmtablette 10 mg / 20 mg / 30 mg), Croscarmellose-Natrium (entspricht 0,56 mg / 1,12 mg / 1,68 mg pro Filmtablette 10 mg / 20 mg / 30 mg Natrium), Magnesiumstearat.
Filmüberzug: Titandioxid E171, Poly(vinylalkohol), Macrogol, Talkum, Eisenoxid rot E172 (bei Filmtabletten 10 mg, 20 mg und 30 mg), Eisenoxid gelb E172 (nur bei Filmtabletten 20 mg und 30 mg), Eisenoxid schwarz E172 (nur bei Filmtabletten 30 mg).
Indikationen/AnwendungsmöglichkeitenPsoriasis
Otezla ist indiziert zur Behandlung von erwachsenen Patienten mit mittelschwerer bis schwerer Plaque-Psoriasis, die auf eine andere systemische Therapie nicht angesprochen haben, eine solche nicht tolerieren oder wenn eine solche kontraindiziert ist.
Psoriatische Arthritis
Otezla ist als Monotherapie oder in Kombination mit krankheitsmodifizierenden Antirheumatika (DMARDs) zur Behandlung der aktiven Psoriasis-Arthritis bei erwachsenen Patienten indiziert, die auf eine vorhergehende Therapie mit DMARDs nicht angesprochen haben oder eine solche nicht tolerieren oder wenn eine solche kontraindiziert ist.
Morbus Behçet
Otezla ist indiziert für die Behandlung persistierender, mit Morbus Behçet assoziierter oraler Ulcera bei erwachsenen Patienten, die ungenügend auf eine topische Therapie angesprochen haben.
Dosierung/AnwendungDie empfohlene Dosierung für Patienten mit Psoriasis, Psoriatischer Arthritis und Morbus Behçet beträgt 30 mg zweimal täglich, morgens und abends im Abstand von etwa 12 Stunden, oral unter Anwendung eines initialen Titrationsschema wie in der nachfolgenden Tabelle dargestellt.
Dosistitrationsschema
|
Tag 1
|
Tag 2
|
Tag 3
|
Tag 4
|
Tag 5
|
Ab Tag 6
| |
Morgens
|
Abends
|
Morgens
|
Abends
|
Morgens
|
Abends
|
Morgens
|
Abends
|
Morgens
|
Abends
|
Morgens
|
Abends
| |
10 mg
|
Keine Dosis
|
10 mg
|
10 mg
|
10 mg
|
20 mg
|
20 mg
|
20 mg
|
20 mg
|
30 mg
|
30 mg
|
30 mg
|
Psoriasis, Psoriatische Arthritis
In zulassungsrelevanten Studien wurde die grösste Verbesserung innerhalb der ersten 24 Behandlungswochen beobachtet. Ist bei einem Patienten nach 24 Wochen noch kein therapeutischer Nutzen erkennbar, sollte die Behandlung überdacht werden.
Abgesehen von nicht-biologischen DMARDs in Psoriatischer Arthritis wurde die Kombinationstherapie mit anderen systemischen Therapien sowie die Kombination mit PUVA nicht untersucht.
Die Kombinationstherapie mit Biologika wurde nicht untersucht und wird aufgrund des theoretischen Risikos für eine Kumulation unerwünschter Wirkungen nicht empfohlen.
Morbus Behçet
In der zulassungsrelevanten Studie wurde die grösste Verbesserung innerhalb der ersten 12 Behandlungswochen beobachtet. Ist bei einem Patienten nach 12 Wochen noch kein therapeutischer Nutzen erkennbar, sollte die Behandlung überdacht werden.
Klinische Daten über 64 Wochen hinaus liegen nicht vor.
Die Kombinationstherapie mit anderen systemischen immunmodulatorischen Therapien, einschliesslich Biologika, wurde nicht untersucht und wird aufgrund des Risikos für eine Kumulation unerwünschter Wirkungen nicht empfohlen.
Art der Anwendung
Die Otezla Filmtabletten sind unzerkaut zu schlucken und können zu den Mahlzeiten oder unabhängig davon eingenommen werden. Die Tabletten dürfen nicht zerstossen, zerteilt oder zerkaut werden.
Spezielle Dosierungsanweisungen
Pädiatrie (<18 Jahre)
Die Sicherheit und Wirksamkeit von Otezla bei Kindern und Jugendlichen unter 18 Jahren wurde nicht untersucht.
Ältere Patienten
Eine Dosisanpassung ist bei Otezla nicht erforderlich.
Die Erfahrung mit Morbus Behçet Patienten über 65 Jahre ist limitiert.
Patienten mit Nierenfunktionsstörungen
Bei Patienten mit leichter und mässiger Niereninsuffizienz ist keine Dosisanpassung erforderlich. Bei Patienten mit mässiger Niereninsuffizienz liegen nur begrenzt Daten vor.
Bei Patienten mit schwerer Einschränkung der Nierenfunktion (CLcr <30 ml/min, berechnet nach der Cockcroft-Gault-Formel) soll die Dosierung von Otezla auf 30 mg einmal täglich reduziert werden. Für die initiale Dosistitration wird in dieser Patientengruppe empfohlen, Apremilast nur mit den im obigen Dosistitrationsschema angegebenen Morgendosen zu titrieren und die Abenddosen auszulassen. Es liegen keine Daten für Dialysepatienten vor. Die Anwendung von Apremilast wird bei dieser Patientengruppe nicht empfohlen (siehe «Pharmakokinetik»).
KontraindikationenÜberempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe.
Schwangerschaft (siehe «Schwangerschaft/Stillzeit»).
Warnhinweise und VorsichtsmassnahmenDosierung bei Niereninsuffizienz
Bei Patienten mit schwer eingeschränkter Nierenfunktion muss die Dosis auf einmal 30 mg Otezla pro Tag reduziert werden. Die Anwendung bei Dialysepatienten wird nicht empfohlen.
Depressionen
Die Behandlung mit Otezla kann mit Depressionen als Nebenwirkung verbunden sein. Bevor Otezla bei Patienten mit Depressionen und/oder suizidalen Gedanken oder suizidalem Verhalten in der Vorgeschichte angewendet wird, soll eine sorgfältige Nutzen-Risiko-Abwägung durchgeführt werden. Die Patienten, ihre Betreuungspersonen und Angehörigen sind darauf hinzuweisen, dass auf das Auftreten bzw. eine Verschlechterung von Depressionen, suizidalen Gedanken oder sonstige Veränderungen der Stimmungslage unbedingt zu achten und beim Auftreten solcher Veränderungen der Arzt unmittelbar zu kontaktieren ist.
Beim Auftreten solcher Ereignisse soll eine sorgfältige Nutzen-Risiko-Abwägung im Hinblick auf eine Fortsetzung der Behandlung mit Otezla vorgenommen werden.
Vorbestehende Infektionen
Otezla wurde bei Patienten mit aktiven bakteriellen Infektionen oder HCV-, HBV- und HIV-Infektionen nicht untersucht und soll daher bei diesen Patienten nicht angewendet werden.
Durchfall, Übelkeit und Erbrechen
Nach der Marktzulassung gab es Berichte über schweren Durchfall, Übelkeit und Erbrechen, die mit der Einnahme von Otezla in Verbindung gebracht wurden. Die meisten Ereignisse traten in den ersten Behandlungswochen auf. In einigen Fällen wurden die Patienten hospitalisiert. Patienten, die 65 Jahre oder älter sind und Patienten die Medikamente zur Volumenreduzierung oder Hypotonie einnehmen, können ein erhöhtes Komplikationsrisiko haben. Patienten, die die Dosis reduzierten oder Otezla abgesetzt haben, erholten sich schnell.
Wenn Patienten eine schwere Form von Diarrhoe, Übelkeit oder Erbrechen entwickeln, kann ein Absetzen der Behandlung mit Otezla erforderlich sein.
Patienten mit Morbus Behçet und systemischen Manifestationen
Patienten mit Morbus Behçet und aktiver Beteiligung wichtiger Organe (z.B. Manifestationen im Bereich der Augen, des kardiovaskulären Systems, des Gastrointestinaltrakts oder des Zentralnervensystems), bei denen deshalb im Jahr vor dem Screening eine immunsuppressive Therapie erforderlich war, wurden aus der Studie ausgeschlossen.
Unverträglichkeiten
Otezla Filmtabletten enthalten Lactose. Patienten mit den seltenen hereditären Stoffwechselstörungen wie Galactose-Intoleranz, Lapp-Lactase-Mangel oder Glucose-Galactose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Dieses Arzneimittel enthält weniger als 1 mmol Natrium (23 mg) pro Filmtablette, d.h. es ist nahezu «natriumfrei».
InteraktionenDie Co-Administration von Otezla mit mehreren Dosen Rifampicin resultierte in einer Abnahme der AUC und der maximalen Serumkonzentration (Cmax) von Apremilast um ca. 72% resp. 43%. Die Exposition gegenüber Apremilast nimmt bei gleichzeitiger Verabreichung von starken CYP3A4-Induktoren (z.B. Rifampicin) ab und kann zu einem geringeren klinischen Ansprechen führen. Die gleichzeitige Anwendung starker Cytochrom P450 Induktoren wie beispielsweise Rifampicin, Phenobarbital, Carbamazepin, Phenytoin und Johanniskraut mit Otezla wird daher nicht empfohlen.
Die gleichzeitige Verabreichung von Ketoconazol bewirkte einen Anstieg der mittleren AUC0-∞- und Cmax-Werte von Apremilast um ca. 36% resp. 5%, was klinisch nicht bedeutsam ist. Otezla kann zusammen mit einem potenten CYP3A4-Inhibitor wie Ketoconazol verabreicht werden.
Es bestanden keine pharmakokinetische Arzneimittelinteraktionen zwischen Apremilast 30 mg zweimal täglich und Methotrexat sowohl bei Patienten mit rheumatoider Arthritis (N=12), welche MTX als Einzeldosis von 10 mg, 12,5 mg, 15 mg, 17,5 mg und 20 mg erhielten als auch nicht bei Psoriasis-Arthritis Patienten (N=3), welche stabile orale MTX Dosen (zwischen 7,5 mg und 20 mg während mindestens 3 Monaten) erhielten.
Zwischen Apremilast und oralen Kontrazeptiva, welche Ethinylestradiol und Norgestimat enthalten, bestand keine pharmakokinetische Interaktion. Otezla kann zusammen mit oralen Kontrazeptiva eingenommen werden.
Schwangerschaft, StillzeitFrauen im gebärfähigen Alter
Während der Behandlung mit Apremilast sollen sichere Empfängnisverhütungsmethoden angewendet werden.
Schwangerschaft
Es liegen keine hinreichenden und gut kontrollierten Studien zur Anwendung von Apremilast bei Schwangeren vor. Apremilast ist während der Schwangerschaft kontraindiziert.
Bei Apremilast Dosen, welche die zurzeit höchste für den Menschen empfohlene Dosis überschritten, wurden Effekte wie embryofötaler Verlust bei Mäusen und Affen, sowie verminderte Fötengewichte und verzögerte Ossifikation bei Mäusen beobachtet. Keine solchen Effekte wurden beobachtet, wenn die Exposition dem 1,3-Fachen der klinischen Exposition entsprach (siehe «Präklinische Daten»).
Stillzeit
Apremilast wurde in der Milch laktierender Mäuse nachgewiesen. Es ist nicht bekannt, ob Apremilast oder seine Metaboliten beim Menschen in die Muttermilch übergehen.
Frauen, die mit Apremilast behandelt werden, sollen nicht stillen.
Wirkung auf die Fahrtüchtigkeit und auf das Bedienen von MaschinenOtezla hat keinen Einfluss auf die Fahrtüchtigkeit oder die Fähigkeit, Maschinen zu bedienen.
Unerwünschte WirkungenDie in den klinischen Phase-3-Studien zu PsA und PSOR (Studien PALACE 1, PALACE 2, PALACE 3, PALACE 4 sowie ESTEEM 1 und ESTEEM 2) am häufigsten gemeldeten Nebenwirkungen waren gastrointestinale (GI) Störungen einschliesslich Durchfall (15,7%) und Übelkeit (13,9%). Diese GI-Nebenwirkungen waren meist leicht bis mässig ausgeprägt, wobei 0,3% der Patienten über schwere Durchfälle und 0,3% der Patienten über starke Übelkeit berichteten. Diese Nebenwirkungen traten im Allgemeinen innerhalb der ersten 2 Wochen der Behandlung auf und bildeten sich in der Regel innerhalb von 4 Wochen wieder zurück. Zu den weiteren am häufigsten berichteten Nebenwirkungen gehörten Infektionen der oberen Atemwege (8,4%), Kopfschmerzen (7,9%) und Spannungskopfschmerzen (7,2%). Insgesamt wurden die meisten Nebenwirkungen als leicht oder mässig eingestuft.
Die häufigsten Nebenwirkungen, die in den Phase-3-Studien zu PsA und PSOR in den ersten 16 Wochen der Behandlung zum Therapieabbruch führten, waren Durchfall (1,7%) und Übelkeit (1,5%). Die Inzidenz schwerwiegender Nebenwirkungen war gering und liess nicht erkennen, dass ein bestimmtes Organsystem besonders betroffen wäre.
Die Langzeitsicherheit von Apremilast 30 mg zweimal täglich bei Patienten mit psoriatischer Arthritis und Psoriasis wurde für eine Gesamtbehandlungsdauer von bis zu 5 Jahren untersucht. Die Langzeiterfahrung in unverblindeten Verlängerungsstudien mit Apremilast war mit den 52-Wochen Studien vergleichbar.
Die am häufigsten gemeldeten unerwünschten Arzneimittelwirkungen von Apremilast bei Morbus Behçet waren Durchfall (41,3%), Übelkeit (19,2%), Kopfschmerz (14,4%), Infektion der oberen Atemwege (11,5%), Oberbauchschmerzen (8,7%), Erbrechen (8,7%) und Rückenschmerzen (7,7%), und diese waren überwiegend von leichtem bis mittlerem Schweregrad.
Die gastrointestinalen Nebenwirkungen traten im Allgemeinen innerhalb der ersten zwei Wochen der Behandlung auf und bildeten sich in der Regel innerhalb von vier Wochen wieder zurück.
Die Nebenwirkungen sind unten nach Systemorganklasse (MedDRA SOC) und Häufigkeit aufgeführt. Innerhalb jeder SOC und Häufigkeitskategorie sind die Nebenwirkungen nach absteigender Bedeutung aufgeführt.
Die unerwünschten Arzneimittelwirkungen wurden anhand von Daten aus den Phase-3-Studien des klinischen Entwicklungsprogramms von Apremilast ermittelt. Bei den angegebenen Häufigkeiten der unerwünschten Arzneimittelwirkungen handelt es sich um die Häufigkeiten in den Otezla-Armen der vier Phase-3-Studien bei PsA (n=1945) resp. der beiden Phase-3-Studien bei PSOR (n=1184) (angegeben ist jeweils die höhere Häufigkeit in den beiden Datenbeständen) und in der Phase-3-Studie zu Morbus Behçet (n=207).
Die Häufigkeitsangaben sind wie folgt definiert: sehr häufig (≥1/10), häufig (≥1/100, <1/10), gelegentlich (≥1/1000, <1/100), selten (≥1/10'000, <1/1000), sehr selten (<1/10'000).
Infektionen und parasitäre Erkrankungen
Häufig: Bronchitis, Infektion der oberen Atemwege, Nasopharyngitis.
Erkrankungen des Immunsystems
Gelegentlich: Überempfindlichkeit.
Stoffwechsel und Ernährungsstörungen
Häufig: Verminderter Appetit.
Psychiatrische Erkrankungen
Häufig: Schlafstörungen, Depressionen.
Erkrankungen des Nervensystems
Häufig: Migräne, Spannungskopfschmerzen, Kopfschmerzen.
Herzerkrankungen
Gelegentlich: Tachyarrhythmie
Erkrankungen der Atemwege, des Brustraums und Mediastinums
Häufig: Husten.
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Durchfall (15,7%), Übelkeit (13,9%).
Häufig: Erbrechen, häufiger Stuhlgang, Oberbauchschmerzen, gastroösophageale Refluxkrankheit, Dyspepsie.
Erkrankungen der Haut und des Unterhautgewebes
Gelegentlich: Hautausschlag.
Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen
Häufig: Rückenschmerzen.
Allgemeine Erkrankungen und Beschwerden am Verabreichungsort
Häufig: Fatigue.
Gelegentlich: Gewichtsabnahme.
Weitere Informationen
Morbus Behçet
In der Studie zu M. Behçet wurde das Sicherheitsprofil von Otezla grundsätzlich bestätigt (siehe obige Liste). Einige unerwünschte Wirkungen wurden häufiger beobachtet in der Behçet Studie.
Infektionen und parasitäre Erkrankungen
Sehr häufig: Infektion der oberen Atemwege (11,5%).
Erkrankungen des Nervensystems
Sehr häufig: Kopfschmerzen (14,4%).
Erkrankungen des Gastrointestinaltrakts
Sehr häufig: Durchfall (41,3%), Übelkeit (19,2%).
Überempfindlichkeitsreaktionen
Die Patienten mit bestätigter Überempfindlichkeit berichteten über Engegefühl des Halses, Pruritus, Urtikaria, Hautquaddeln und Ausschlag.
Gewichtsabnahme
Das Gewicht der Patienten wurde in den klinischen Studien routinemässig kontrolliert. Bis zur 16. Behandlungswoche wurde Gewichtsabnahme als UAW bei 9 Patienten (0,8%) der mit Apremilast 30 mg zweimal täglich behandelten Gruppe und bei 1 Patienten (0,2%) der Placebo-Gruppe berichtet. Nach 52 Wochen betrug der beobachtete Gewichtsverlust im Median bei den Patienten, die mit Otezla 30 mg zweimal täglich behandelt wurden, 1,40 kg. Insgesamt hatten 19,2% der Patienten, die Apremilast erhielten, einen Gewichtsverlust von mehr als 5% beobachtet. Bei keinem Patienten wurde über Gewichtsabnahme als schwerwiegende UAW berichtet. Insgesamt 2 (0,2%) der mit Apremilast behandelten Patienten setzten die Behandlung wegen der Nebenwirkung Gewichtsabnahme ab. Bei keinem Patienten wurden infolge der Gewichtsabnahme manifeste klinische Folgen verzeichnet.
Depression
Während des placebokontrollierten Abschnitts der klinischen Phase-3-Studien zu PSOR berichteten 1,2% (14/1184) der mit Apremilast behandelten im Vergleich zu 0,5% (2/418) der mit Placebo behandelten Patienten über Depressionen. Keine dieser Depressionen wurde als schwerwiegend eingestuft oder führte zum Ausscheiden aus der Studie.
Während des placebokontrollierten Abschnitts der klinischen Phase-3-Studien zu PSA berichteten 0,9% (18/1945) der mit Apremilast behandelten im Vergleich zu 0,7% (5/671) der mit Placebo behandelten Patienten über Depressionen. Depression/depressive Verstimmung wurde in 0,1% (2/1945) der mit Apremilast behandelten und keinem der mit Placebo behandelten Patienten als schwerwiegend gemeldet. Drei (3/1945; 0,2%) der mit Apremilast behandelt Patienten brachen die Studienteilnahme auf Grund von Depressionen oder depressiver Verstimmung ab.
In den Otezla Phase-2- und Phase-3-Studien wurden unter Apremilast jeweils 2 Fälle mit suizidalen Ideen bzw. Suizidversuchen und kein Suizid beobachtet, gegenüber 2 Suiziden unter Placebo.
Weitere spezielle Patientengruppen
Sicherheit bei älteren Patienten
In den klinischen Studien wurden beim Sicherheitsprofil insgesamt keine Unterschiede zwischen älteren Patienten ab 65 Jahren und jüngeren erwachsenen Patienten unter 65 Jahren beobachtet.
Patienten mit Leberfunktionsstörungen
Die Sicherheit von Otezla wurde bei Patienten mit PsA-, PSOR- oder Morbus Behçet mit eingeschränkter Leberfunktion nicht untersucht.
Patienten mit eingeschränkter Nierenfunktion
In den klinischen Studien zu PsA, PSOR oder Morbus Behçet war das bei Patienten mit leichter Niereninsuffizienz beobachtete Sicherheitsprofil vergleichbar mit dem von Patienten mit normaler Nierenfunktion. In den klinischen Studien wurde die Sicherheit von Otezla bei Patienten mit PsA-, PSOR- oder Morbus Behçet mit mässiger oder schwerer Niereninsuffizienz nicht untersucht.
Die Meldung des Verdachts auf Nebenwirkungen nach der Zulassung ist von grosser Wichtigkeit. Sie ermöglicht eine kontinuierliche Überwachung des Nutzen-Risiko-Verhältnisses des Arzneimittels. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdacht einer neuen oder schwerwiegenden Nebenwirkung über das Online-Portal ElViS (Electronic Vigilance System) anzuzeigen. Informationen dazu finden Sie unter www.swissmedic.ch.
ÜberdosierungOtezla wurde bei gesunden Probanden in einer maximalen Tagesgesamtdosis von 100 mg (50 mg zweimal täglich) über 4,5 Tage untersucht, ohne dass sich ein Anhaltspunkt für dosislimitierende Toxizitäten ergab. Bei Überdosierung wird empfohlen, den Patienten auf Anzeichen und Symptome von unerwünschten Wirkungen zu überwachen und eine entsprechende symptomatische Behandlung einzuleiten. Im Falle einer Überdosierung sollten die Patienten symptomatisch und unterstützend behandelt werden.
Eigenschaften/WirkungenATC-Code
L04AA32
Wirkungsmechanismus
Apremilast ist ein oraler kleinmolekularer Inhibitor der Phosphodiesterase 4 (PDE4). PDE4 ist eine für zyklisches Adenosinmonophosphat (cAMP) spezifische und in Entzündungszellen wichtige PDE. Durch PDE4-Hemmung werden intrazelluläre cAMP-Spiegel angehoben. Die spezifischen Mechanismen, über welche die Psoriasis/Psoriasis-Arthritis und der Morbus Behçet beeinflusst werden, sind nicht vollständig aufgeklärt.
Pharmakodynamik
In klinischen Studien an Patienten mit Psoriasis-Arthritis bewirkte Apremilast eine signifikante Modulation, jedoch keine vollständige Hemmung der Plasmaproteinspiegel von IL-1α, IL-6, IL-8, MCP-1, MIP-1β, MMP-3 und TNF-α. Nach 40-wöchiger Behandlung mit Apremilast fand sich eine Abnahme der Plasmaproteinspiegel von IL-17 und IL-23 sowie ein Anstieg von IL-10. In klinischen Studien an Psoriasis-Patienten verminderte Apremilast die Epidermisdicke der von Läsionen befallenen Haut, die Infiltration durch Entzündungszellen und die Expression proinflammatorischer Gene, einschliesslich derjenigen für induzierbare Stickoxidsynthase (iNOS), IL-12/IL-23p40, IL-17A, IL-22 und IL-8. Bei Patienten mit Morbus Behçet, die mit Apremilast behandelt wurden, bestand ein positiver Zusammenhang zwischen der Veränderung des Plasma-TNF-alpha und der klinischen Wirksamkeit, gemessen anhand der Anzahl von oralen Ulcera.
Bei therapeutischen Plasmakonzentrationen führte Apremilast bei gesunden Probanden zu keiner Verlängerung des QT-Intervalls.
Klinische Wirksamkeit
Psoriasis-Arthritis
Erfahrungen aus klinischen Studien an Patienten mit Psoriasis-Arthritis, welche mit kleinmolekularer DMARDs und/oder Biologika vortherapiert waren
Die Sicherheit und Wirksamkeit von Otezla wurde in 3 ähnlich aufgebauten multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien (Studien PALACE 1, PALACE 2 und PALACE 3) an 1493 erwachsenen Patienten mit aktiver PsA (≥3 geschwollene Gelenke und ≥3 druckschmerzhafte Gelenke) trotz Vortherapie mit DMARDs, einschliesslich biologischer DMARDs (z.B. TNF-Blocker), oder bestehender kleinmolekularer DMARD-Therapie untersucht.
Bei den Patienten in diesen Studien bestand die Diagnose PsA seit mindestens 6 Monaten. In der Studie PALACE 3 war zudem eine qualifizierende Psoriasis-Hautläsion (Mindestdurchmesser 2 cm) erforderlich. Patienten, bei denen >3 bei PsA eingesetzte Substanzen (kleine Moleküle oder Biologika) oder >1 biologischer TNF-Blocker bereits versagt hatten, wurden ausgeschlossen. In die 3 Studien wurden Patienten mit allen PsA-Unterformen eingeschlossen: symmetrische Polyarthritis (62,0%), asymmetrische Oligoarthritis (26,9%), Arthritis mit Befall der distalen Interphalangealgelenke (DIP) (6,2%), Arthritis mutilans (2,7%) und prädominante Spondylitis (2,1%). Patienten mit vorbestehender Enthesitis (63%) und vorbestehender Daktylitis (42%) wurden aufgenommen.
In den 3 Studien wurden die Patienten zu Placebo (n=496), Otezla 20 mg (n=500) oder Otezla 30 mg (n=497) zweimal täglich oral randomisiert. In den Studien PALACE 1, PALACE 2 und PALACE 3 erfolgte die Behandlungszuordnung stratifiziert nach bestehender Therapie mit kleinmolekularen DMARDs in der Ausgangslage (Baseline). In PALACE 3 war ein Psoriasisbefall von ≥3% der Körperoberfläche (KOF) ein weiteres Stratifizierungskriterium. Die Patienten durften während der Studie eine Begleittherapie mit stabilen Dosen von Methotrexat (MTX) (≤25 mg/Woche; 54,5%), Sulfasalazin (SSZ) (≤2 g/Tag; 9,0%), Leflunomid (LEF) (≤20 mg/Tag; 7,4%), niedrig dosierten oralen Kortikosteroiden (entsprechend ≤10 mg Prednison täglich; 13,9%) und/oder nichtsteroidalen Antirheumatika (NSAR; 70,7%) erhalten. Die Gabe von Apremilast in Kombination mit biologischen DMARDs wurde nicht untersucht.
Eine Vortherapie nur mit kleinmolekularen DMARDs wurde bei 76,4% der Patienten und eine Vortherapie mit biologischen DMARDs bei 22,4% der Patienten angegeben, darunter 7,8% mit Versagen einer biologischen DMARD-Vortherapie. Die mediane PsA-Erkrankungsdauer betrug 5 Jahre.
Primärer Endpunkt war der Anteil der Patienten, die nach 16 Wochen ein American College of Rheumatology (ACR) 20-Ansprechen erreichten. Patienten, deren Anzahl an druckschmerzhaften und geschwollenen Gelenken keine mindestens 20%ige Besserung aufwiesen, wurden nach 16 Wochen als Nonresponder eingestuft. Placebo-Nonresponder wurden im Verhältnis 1:1 zu Otezla 20 mg zweimal täglich oder 30 mg zweimal täglich verblindet rerandomisiert. Otezla-Patienten erhielten weiterhin ihre initiale Behandlung. Nach 24 Wochen wurden alle verbliebenen Placebo-Patienten entweder zu Otezla 20 mg zweimal täglich oder zu Otezla 30 mg zweimal täglich rerandomisiert. Im Anschluss an die 52 Wochen Behandlung konnten die Patienten in den Langzeit-Verlängerungsstudien der PALACE 1, PALACE 2 und PALACE 3 Studien für insgesamt bis zu 5 Jahre (260 Wochen) unverblindet mit Apremilast 20 mg oder 30 mg fortfahren.
Der Anteil der Patienten, welche ein ACR-20/50/70 Ansprechen in der Studien PALACE 1/2/3 nach 16, 24 und 52 Wochen zeigten, ist in Tabelle 1 aufgelistet. Die Behandlung mit Apremilast führte zu einer signifikanten Verbesserung der Anzeichen und Symptome der PsA, welche anhand der ACR20-Ansprechkriterien bis Woche 16 im Vergleich zu Placebo erhoben wurden. Das ACR-20/50/70 Ansprechen wurden bis Woche 24 aufrechterhalten. Bei den Patienten, welche diejenige Apremilast-Behandlung durchgehend erhielten, auf die sie zu Studienbeginn randomisiert worden waren, wurde das ACR-20/50/70 Ansprechen bis einschliesslich Woche 52 aufrechterhalten. Von den 497 Patienten, welche anfänglich zu Apremilast 30 mg zweimal täglich randomisiert wurden, traten 375 (75%) in die Langzeit-Verlängerungsstudien ein und von diesen waren nach 260 Wochen noch 221 Patienten (59%) unter Behandlung. Das ACR Ansprechen wurde in den Langzeit-Verlängerungsstudien für bis zu 5 Jahre aufrechterhalten.
Tabelle 1: Anteil der Patienten mit ACR-20/50/70 Ansprechen in den Studien PALACE 1/2/3 nach 16, 24 und 52 Wochen
|
|
PALACE 1
|
PALACE 2
|
PALACE 3
| |
|
Placebo
|
Otezla 30 mg zweimal täglich
|
Placebo
|
Otezla 30 mg zweimal täglich
|
Placebo
|
Otezla 30 mg zweimal täglich
| |
Na
|
%
|
Na
|
%
|
Na
|
%
|
Na
|
%
|
Na
|
%
|
Na
|
%
| |
ACR 20
| |
Woche 16
|
168
|
19,0%
|
168
|
38,1%***
|
159
|
18,9%
|
162
|
32,1%**
|
169
|
18,3%
|
167
|
40,7%***
| |
Woche 24
|
168
|
13,1%
|
168
|
35,1%***
|
159
|
15,7%
|
162
|
24,7%*
|
169
|
15,4%
|
167
|
31,1%***
| |
Woche 52a
|
N/Ab
|
130
|
54,6%
|
N/Ab
|
116
|
52,6%
|
N/Ab
|
127
|
63,0%
| |
ACR 50
| |
Woche 16
|
168
|
6,0%
|
168
|
16,1%**
|
159
|
5,0%
|
162
|
10,5%
|
169
|
8,3%
|
167
|
15,0%
| |
Woche 24
|
168
|
4,2%
|
168
|
19,0%***
|
159
|
8,8%
|
162
|
11,7%
|
169
|
7,7%
|
167
|
16,2%*
| |
Woche 52a
|
N/Ab
|
130
|
24,6%
|
N/Ab
|
118
|
18,6%
|
N/Ab
|
126
|
30,2%
| |
ACR 70
| |
Woche 16
|
168
|
1,2%
|
168
|
4,2%
|
159
|
0,6%
|
162
|
1,2%
|
169
|
2,4%
|
167
|
3,6%
| |
Woche 24
|
168
|
0,6%
|
168
|
10,1%***
|
159
|
3,1%
|
162
|
2,5%
|
169
|
3,6%
|
167
|
5,4%
| |
Woche 52a
|
N/Ab
|
130
|
13,8%
|
N/Ab
|
118
|
6,8%
|
N/Ab
|
125
|
10,4%
|
N/A=nicht zutreffend.
* p ≤0,05 verglichen mit Placebo; ** p ≤0,01 verglichen mit Placebo; *** p ≤0,001 verglichen mit Placebo
a Die Ansprechraten über 16 und 24 Wochen bezieht sich auf N=Anzahl der randomisierten Patienten, die Ansprechrate bei Woche 52 bezieht sich auf N=Anzahl der bis zu diesem Zeitpunkt in der Studie verbliebenen Patienten.
b Kein Placebo nach Woche 24.
Die in der mit Apremilast behandelten Gruppe beobachteten Ansprechraten waren bei Patienten mit bzw. ohne Begleittherapie mit DMARDs, einschliesslich MTX, vergleichbar (Tabelle 2). Bei den Zuvor ausschliesslich mit niedermolekularen DMARDs oder Biologika vortherapierten Patienten, die Apremilast erhielten, war das ACR20-Ansprechen bis Woche 16 im Vergleich zu den mit Placebo behandelten Patienten grösser (Tabelle 3).
Ähnliche ACR20-Ansprechraten wurden bei Patienten mit verschiedenen PsA-Unterformen, einschliesslich Arthritis mit Befall der DIP, beobachtet; die Anzahl von Patienten mit den Unterformen Arthritis mutilans und prädominante Spondylitis war jedoch zu gering, um eine sinnvolle Bewertung zu erlauben.
Tabelle 2: Anteil der Patienten mit ACR-20 Ansprechen in den Studien PALACE 1/2/3 nach 16 Wochen mit oder ohne DMARD Begleittherapie
|
|
Placebo
|
Otezla 30 mg zweimal täglich
| |
Na
|
%
|
Na
|
%
| |
Monotherapie
| |
PALACE 1
|
58
|
10,3%
|
62
|
46,8%
| |
PALACE 2
|
46
|
15,2%
|
49
|
22,4%
| |
PALACE 3
|
68
|
13,2%
|
66
|
39,4%
| |
Mit DMARD Begleittherapie
| |
PALACE 1
|
110
|
23,6%
|
106
|
33,0%
| |
PALACE 2
|
113
|
20,4%
|
113
|
36,3%
| |
PALACE 3
|
101
|
21,8%
|
101
|
41,6%
|
a N ist die Anzahl randomisierter und behandelter Patienten innerhalb jeder Subgruppe.
Tabelle 3: Anteil der Patienten, die in PALACE 1, 2 und 3 nach vorherige Anwendung von Biologika in Woche 16 eine ACR-20-Response erreichten
|
Subgruppe
Studie
|
Placebo
|
Otezla 30 mg zweimal täglich
| |
Na
|
%
|
Na
|
%
| |
Vorherige Behandlung nur mit niedermolekularen DMARDs (Biologika-naive Patienten)
| |
PALACE 1
|
120
|
23,3%
|
124
|
41,1%
| |
PALACE 2
|
135
|
20,7%
|
134
|
34,3%
| |
PALACE 3
|
121
|
20,7%
|
124
|
42,7%
| |
Vorherige Behandlung mit Biologikab
| |
PALACE 1
|
41
|
4,9%
|
41
|
26,8%
| |
PALACE 2
|
23
|
8,7%
|
23
|
21,7%
| |
PALACE 3
|
48
|
12,5%
|
43
|
34,9%
|
DMARD=krankheitsmodifizierendes Antirheumatikum.
a N ist die Anzahl der randomisierten und behandelten Patienten innerhalb der jeweiligen Subgruppe.
b Die Teilnehmer konnten vorher mit Biologika und zusätzlich mit niedermolekularen DMARDs behandelt worden sein.
In den Studien PALACE 1, PALACE 2 und PALACE 3 waren die Verbesserungen der Krankheitsaktivitätsskala („Disease Activity Scale“, DAS) 28 mit C-reaktivem Protein (CRP) und der Anteil von Patienten, die ein modifiziertes PsA-Ansprechkriterium (PsARC) erreichten, bis Woche 16 in der Apremilast-Gruppe im Vergleich zu Placebo grösser (nominaler p <0,0004 bzw. p <0,0017). Diese Verbesserungen wurden bis Woche 24 aufrechterhalten. Bei den Patienten, welche diejenige Apremilast-Behandlung durchgehend erhielten, zu der sie zu Studienbeginn randomisiert worden waren, wurden der DAS28(CRP)-Score und das PsARC-Ansprechen bis einschliesslich Woche 52 aufrechterhalten. Die Verzögerung der Progression struktureller Schäden wurde nicht untersucht.
Bis Woche 16 und 24 wurden bei den mit Apremilast behandelten Patienten Verbesserungen bei den für die Psoriasis-Arthritis charakteristischen Parametern der peripheren Krankheitsaktivität (Anzahl geschwollener Gelenke, Anzahl (druck)schmerzempfindlicher Gelenke) und den Hautmanifestationen der Psoriasis beobachtet. Bei den Patienten, welche diejenige Apremilast-Behandlung durchgehend erhielten, zu der sie zu Studienbeginn randomisiert worden waren, wurden diese Verbesserungen bis einschliesslich Woche 52 aufrechterhalten. Das klinische Ansprechen wurde in den Langzeit-Verlängerungsstudien für dieselben Parameter der peripheren Krankheitsaktivität und der Hautmanifestationen der Psoriasis für bis zu 5 Jahre aufrechterhalten.
Körperliche Funktion und Quality of Life
Mit Apremilast 30 mg zweimal täglich behandelte Patienten wiesen eine im Vergleich zur Placebo-Gruppe signifikant grössere Verbesserung bei der mittleren Veränderung des HAQ-DI-Scores gegenüber Baseline nach 16 Wochen und nach 24 Wochen auf. Das Verhältnis der HAQ-DI-Responder (Verbesserung ≥0,3 gegenüber Baseline) nach 16 Wochen war in der 30 mg Otezla Gruppe grösser als in der Placebo-Gruppe (Tabelle 4). Bei den Patienten, welche durchgehend mit Apremilast therapiert wurden, wurde die Verbesserung im HAQ-DI-Score und im Verhältnis zu Placebo über 52 Wochen aufrechterhalten.
Tabelle 4: Veränderung des HAQ-DI-Scores gegenüber der Baseline in den Studien PALACE 1/2/3 in Woche 16, 24 und 52
|
|
Placebo
|
Otezla 30 mg zweimal täglich
| |
Na
|
Änderung
|
Na
|
Änderung
| |
PALACE 1
| |
Woche 16
|
168
|
-0,086
|
168
|
-0,244**
| |
Woche 24
|
168
|
-0,076
|
168
|
-0,258***
| |
Woche 52
|
N/Ab
|
132
|
-0,318
| |
PALACE 2
| |
Woche 16
|
159
|
-0,053
|
162
|
-0,193**
| |
Woche 24
|
159
|
-0,085
|
162
|
-0,206*
| |
Woche 52
|
N/Ab
|
117
|
-0,330
| |
PALACE 3
| |
Woche 16
|
169
|
-0,065
|
167
|
-0,192**
| |
Woche 24
|
169
|
-0,053
|
167
|
-0,192**
| |
Woche 52
|
N/Ab
|
127
|
-0,350
|
* p ≤0,05 für Apremilast vs. Placebo; **p ≤0,01 für Apremilast vs. Placebo; ***p ≤0,001 für Apremilast vs. Placebo.
a Die Ansprechraten über 16 und 24 Wochen bezieht sich auf N=Anzahl der randomisierten Patienten, die Ansprechrate bei Woche 52 bezieht sich auf N=Anzahl der bis zu diesem Zeitpunkt in der Studie verbliebenen Patienten.
b Kein Placebo nach Woche 24.
HAQ-DI=Health Assessment Questionnaire-Disability Index; 0=beste Bewertung; 3=schlechteste Bewertung; mit diesem Fragebogen wird die Fähigkeit des Patienten, folgende Tätigkeiten zu verrichten, ermittelt: Ankleiden/Körperpflege, Aufstehen, Essen, Gehen, Erreichen von Gegenständen, Greifen, Hygiene und Verrichtung von Aktivitäten des täglichen Lebens.
In den Studien PALACE 1, PALACE 2 und PALACE 3 wurden bei den mit Apremilast behandelten Patienten im Vergleich zu Placebo signifikante Verbesserungen der gesundheitsbezogenen Lebensqualität nachgewiesen, erfasst anhand der gegenüber Baseline erhobenen Veränderungen in Gesundheitsfragebogen «Short Form Health Survey» Version 2 (SF-36v2) und im Score des Instruments Funktionelle Beurteilung der Therapie chronischer Erkrankung – Fatigue (FACIT-Fatigue). Eine verbesserte physische Funktion, bewertet gemäss HAQ-DI und SF-36v2PF, sowie der FACIT Fatigue Score wurden in den Langzeit-Verlängerungsstudien für bis zu 5 Jahre aufrechterhalten.
Psoriasis
Die Sicherheit und Wirksamkeit von Otezla wurden in zwei multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studien (Studien ESTEEM 1 und ESTEEM 2) beurteilt, in welche insgesamt 1257 Patienten ab 18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis eingeschlossen wurden, bei denen ≥10% der Körperoberfläche (KOF) befallen war, ein PASI (Psoriasis Area and Severity Index)-Score ≥12 und ein sPGA (static Physician Global Assessment)-Score ≥3 (mittelschwer oder schwer) vorlagen und die für eine Phototherapie oder systemische Therapie in Frage kamen.
Diese Studien waren bis zur Woche 32 ähnlich aufgebaut. In beiden Studien wurden die Patienten für eine 16-wöchige Behandlung im Verhältnis 2:1 zu Otezla 30 mg zweimal täglich oder Placebo randomisiert (placebokontrollierte Phase), und von Woche 16 bis 32 erhielten alle Patienten Otezla 30 mg zweimal täglich (Erhaltungsphase). Während der randomisierten Therapie-Absetzphase (Woche 32 bis 52) wurden diejenigen Patienten, die ursprünglich zu Otezla randomisiert worden waren und eine mindestens 75%ige Reduktion ihres PASI-Scores (PASI-75) (ESTEEM 1) bzw. eine mindestens 50%ige Reduktion ihres PASI-Scores (PASI-50) (ESTEEM 2) erreichten, nach 32 Wochen entweder zu Placebo oder zu Otezla 30 mg zweimal täglich rerandomisiert. Patienten, die zu Placebo rerandomisiert wurden und nach 32 Wochen ihr PASI-75-Ansprechen (ESTEEM 1) bzw. 50% ihrer gegenüber Baseline verzeichneten PASI-Verbesserung (ESTEEM 2) einbüssten, wurden erneut mit Otezla 30 mg zweimal täglich behandelt. Patienten, welche das vorgegebene PASI-Ansprechen bis Woche 32 nicht erreichten oder die anfangs zu Placebo randomisiert worden waren, erhielten Otezla bis Woche 52. Die Gabe von Apremilast in Kombination mit biologischen DMARDs wurde nicht untersucht.
Im Anschluss an die 52 Wochen Behandlung konnten die Patienten in den Langzeit-Verlängerungsstudien der ESTEEM 1 und ESTEEM 2 Studien für insgesamt bis zu 5 Jahre (260 Wochen) unverblindet mit Apremilast 30 mg fortfahren.
Primärer Endpunkt war in beiden Studien der Anteil von Patienten, die nach 16 Wochen ein PASI-75-Ansprechen erreichten. Der wichtigste sekundäre Endpunkt war der Anteil von Patienten, die nach 16 Wochen einen sPGA-Score von befallsfrei (0) oder nahezu befallsfrei (1) erreichten. Zu den weiteren Endpunkten gehörten befallene KOF, Pruritus-VAS, Nagelbefall (NAPSI), Kopfhautbefall (ScPGA) und Resultate aus Fragebögen zur Lebensqualität (DLQI und SF-36 MCS).
Über beide Studien hinweg betrachtet, lag das mediane Alter bei 45,8 Jahren (18-83 Jahre). Bei Baseline betrug der mittlere KOF-Befall 25,19% (Median 21,0%), der mittlere PASI-Score 19,07 (Median 16,80) und der Anteil von Patienten mit einem sPGA-Score von 3 (mittelschwer) und 4 (schwer) 70,0% resp. 29,8%. Etwa 30% aller Patienten hatten zur Behandlung der Psoriasis bereits eine Phototherapie und 54% eine konventionelle systemische und/oder biologische Vortherapie erhalten (einschliesslich Therapieversagern), davon 37% eine herkömmliche systemische Vorbehandlung und 30% eine Vortherapie mit Biologika. Etwa ein Drittel der Patienten war weder mit einer Phototherapie noch mit einer konventionellen systemischen oder biologischen Therapie vorbehandelt worden. Bei insgesamt 18% der Patienten war eine Psoriasis-Arthritis in der Vorgeschichte bekannt.
Die Anteile von Patienten mit einem PASI-50-, PASI-75- und PASI-90-Ansprechen und einem sPGA-Score von befallsfrei (0) oder nahezu befallsfrei (1) sind in der nachfolgenden Tabelle (Tabelle 5) dargestellt. Otezla bewirkte eine im Vergleich zu Placebo signifikante Besserung der mittelschweren bis schweren Plaque-Psoriasis, erhoben anhand des Anteils von Patienten mit einem PASI-75-Ansprechen nach 16 Wochen. Auch für das sPGA-, PASI-50- und PASI-90-Ansprechen konnten nach 16 Wochen klinische Verbesserungen belegt werden.
Tabelle 5: Klinisches Ansprechen nach 16 Wochen in den Studien ESTEEM 1 und ESTEEM 2 (FASc; LOCF)
|
|
ESTEEM 1
|
ESTEEM 2
| |
|
Placebo
|
Otezla 30 mg zweimal täglich*
|
Placebo
|
Otezla 30 mg zweimal täglich*
| |
N
|
282
|
562
|
137
|
274
| |
PASIa 75, n (%)
|
15 (5,3)
|
186 (33,1)
|
8 (5,8)
|
79 (28,8)
| |
sPGAb «befallsfrei» oder
«nahezu befallsfrei», n (%)
|
11 (3,9)
|
122 (21,7)
|
6 (4,4)
|
56 (20,4)
| |
PASI 50, n (%)
|
48 (17,0)
|
330 (58,7)
|
27 (19,7)
|
152 (55,5)
| |
PASI 90, n (%)
|
1 (0,4)
|
55 (9,8)
|
2 (1,5)
|
24 (8,8)
|
* p <0,0001 für alle Vergleiche versus Placebo ausser für PASI-90-Ansprechen in der Studie ESTEEM 2 – dort p=0,0042.
a PASI=Psoriasis Area and Severity Index.
b sPGA=Static Physician Global Assessment.
c FAS=Full Analysis Set.
Der klinische Nutzen von Otezla wurde für verschiedene Subgruppen nachgewiesen, welche anhand von demographischen Charakteristika bei Baseline, klinischen Charakteristika der Erkrankung im Ausgangsbefund (einschliesslich Dauer der Psoriasis-Erkrankung und Patienten mit Psoriasis-Arthritis in der Vorgeschichte), Vortherapie mit Psoriasismitteln und Ansprechen auf Psoriasis-Vortherapien definiert wurden. Über alle nach dem Körpergewicht definierten Subgruppen hinweg wurden vergleichbare Ansprechraten beobachtet. Das Ansprechen auf Otezla setzte rasch ein, wobei bereits innert 2 Wochen im Vergleich zu Placebo signifikant grössere Verbesserungen der Anzeichen und Symptome der Psoriasis, einschliesslich PASI, Hautbeschwerden/-schmerzen und Pruritus, verzeichnet wurden. Das PASI-Ansprechen wurde generell innert 16 Wochen erreicht und bis Woche 32 aufrechterhalten.
Während der randomisierten Therapie-Absetzphase (Woche 32-52) in der Studie ESTEEM 1, blieb die mittlere prozentuale Besserung im PASI bei den Pateinten, welche in Woche 32 zu Otezla rerandomisiert wurden, gegenüber Baseline stabil (81–88%). Annähernd 61% dieser Patienten hatten eine PASI-75-Antwort in Woche 52. Von den Patienten welche in Woche 32 zu Placebo rerandomisiert wurden hatten 11,7% eine PASI-75-Antwort in Woche 52. Patienten welche zu Placebo rerandomisiert wurden verloren die PASI-75-Antwort schneller als die Patienten welche zu Otezla rerandomisiert wurden. Die mittlere Zeitdauer bis zum Verlust der PASI-75-Antwort war bei Patienten, welche zu Placebo bzw. zu Otezla rerandomisiert wurden 5,1 bzw. 17,7 Wochen.
In der Studie ESTEEM 2 betrug die mediane Zeit bis zum ersten Verlust der PASI-50-Antwort für Patienten, welche in Woche 32 zu Placebo bzw. zu Otezla rerandomisiert wurden, 12,4 Wochen bzw. 21,9 Wochen.
In der Studie ESTEEM 1 fanden sich nach 16 Wochen bei den Patienten unter Otezla im Vergleich zur Placebo-Gruppe signifikante Verbesserungen (Rückgänge) der Nagelpsoriasis, erhoben anhand der mittleren prozentualen Veränderung des NAPSI (Nail Psoriasis Severity Index) gegenüber Baseline (Otezla 30 mg zweimal täglich: −22,5%; Placebo: +6,5%; p <0,0001). Ähnliche Verbesserungen wurden auch in der Studie ESTEEM 2 beobachtet (Otezla 30 mg zweimal täglich: −29,0%; Placebo: −7,1%, p=0,0052). Weitere Besserungen der Nagelpsoriasis wurden bei Patienten beobachtet, die fortlaufend mit Otezla behandelt wurden, wobei die mittlere prozentuale Veränderung des NAPSI nach 32 Wochen gegenüber Baseline in ESTEEM 1 bei −43,6% und in ESTEEM 2 bei −60,0% lag.
In der Studie ESTEEM 1 fanden sich bei den Patienten unter Otezla im Vergleich zur Placebo-Gruppe signifikante Verbesserungen der Kopfhautpsoriasis mit mindestens mittelschwerer Ausprägung (≥3), erhoben anhand des prozentualen Anteils von Patienten, die nach 16 Wochen einen ScPGA (Scalp Psoriasis Physician's Global Assessment)-Score von befallsfrei (0) oder minimal (1) erreichten (46,5% vs. 17,5%; p <0,0001). Ähnliche Resultate wurden in der Studie ESTEEM 2 beobachtet (Otezla 30 mg zweimal täglich 40,9%; Placebo 17,2%; p <0,0001).
In den Studien ESTEEM 1 und 2 wurden bei den Patienten unter Otezla im Vergleich zur Placebo-Gruppe signifikante Verbesserungen der Lebensqualität, erhoben anhand der Fragebögen DLQI (Dermatology Life Quality Index) und SF-36v2MCS, nachgewiesen.
Von den 832 Patienten, welche anfänglich zu Apremilast 30 mg zweimal täglich randomisiert wurden, traten 443 (53%) in die Langzeit-Verlängerungsstudien von ESTEEM 1 und ESTEEM 2 ein, und von diesen waren nach 260 Wochen noch 115 Patienten (26%) unter Behandlung. Bei Patienten, die in der unverblindeten Verlängerung der ESTEEM 1 und ESTEEM 2 Studien auf Behandlung mit Apremilast blieben, konnten Verbesserungen des PASI-Scores, der befallenen KOF, des Pruritus, des Nagelbefalls sowie der Lebensqualität für bis zu 5 Jahre generell aufrechterhalten werden.
LIBERATE-Studie
Die Sicherheit und Wirksamkeit von Otezla und Etanercept wurden im Rahmen einer multizentrischen, randomisierten, doppelblinden, placebokontrollierten Studie (LIBERATE) untersucht, in welche insgesamt 250 Patienten ab 18 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis aufgenommen wurden, bei denen ein KOF-Befall von ≥10%, ein PASI-Score ≥12 und ein sPGA-Score ≥3 (mittelschwer oder schwer) vorlagen und die für eine Phototherapie oder systemische Therapie in Frage kamen. Ausserdem musste bei den eingeschlossenen Patienten mindestens eine herkömmliche systemische Therapie ein unzureichendes Ansprechen bewirkt haben oder eine Unverträglichkeit gegenüber resp. eine Kontraindikation für eine solche Therapie bestanden haben, und die Studienteilnehmenden durften zudem keine Vortherapie mit Biologika erhalten haben. Die Patienten wurden im Verhältnis 1:1:1 zu Otezla 30 mg oral zweimal täglich, Etanercept 50 mg subkutan einmal wöchentlich oder Placebo für 16 Wochen randomisiert; anschliessend erhielten alle Patienten Otezla 30 mg zweimal täglich. Primärer Endpunkt war das PASI-75-Ansprechen in Woche 16 bei den mit Otezla behandelten Patienten im Vergleich zu Placebo. Ein sekundärer Endpunkt war das PASI-75-Ansprechen bei den mit Etanercept behandelten Patienten im Vergleich zu Placebo. Die Studie war nicht auf die Durchführung statistischer Vergleiche zwischen Otezla und Etanercept ausgelegt, sondern vielmehr auf einen Vergleich jeder Verumbehandlung mit Placebo.
Signifikante Verbesserungen des Anteils von Patienten, welche ein PASI-50, -75 und -90 Ansprechen und einen sPGA-Score von befallsfrei (0) oder nahezu befallsfrei (1) erreichten, fanden sich bei den mit Otezla resp. Etanercept behandelten Patienten jeweils im Vergleich zu Placebo, wie aus untenstehender Tabelle hervorgeht.
Tabelle 6: Klinisches Ansprechen in Woche 16 in der LIBERATE-Studie (mITTa; LOCF)
|
|
Placebo
|
Otezla 30 mg zweimal täglich
|
Etanercept 50 mg einmal wöchentlich
| |
N
|
84
|
83
|
83
| |
PASIb-75, n (%)
|
10 (11,9)
|
33 (39,8)
|
40 (48,2)
| |
[zweiseitiges 95%-KI]c
|
|
[14,9; 40,1]e
|
[23,3; 48,5]e
| |
PASI-50, n (%)
|
28 (33,3)
|
52 (62,7)
|
69 (83,1)
| |
[zweiseitiges 95%-KI]c
|
|
[14,9; 43,9]f
|
[36,9; 62,7]f
| |
PASI-90, n (%)
|
3 (3,6)
|
12 (14,5)
|
17 (20,5)
| |
[zweiseitiges 95%-KI]c
|
|
[2,0; 19,2]g
|
[7,2; 26,1]g
| |
sPGAdScore von befallsfrei (0) oder nahezu befallsfrei (1), n (%)
|
3 (3,6)
|
18 (21,7)
|
24 (28,9)
| |
[zweiseitiges 95%-KI]c
|
|
[8,4; 27,7]h
|
[14,8; 35,5]h
|
a mITT = modifiziertes Intent-to-Treat-Kollektiv.
b PASI = Psoriasis Area and Severity Index.
c Das zweiseitige 95%-Konfidenzintervall (KI) wurde mithilfe des nach BMI stratifizierten CMH-Tests für den Behandlungsunterschied versus Placebo berechnet.
d sPGA=Static Physician Global Assessment.
e Für PASI-75: p <0,0001 für Vergleiche Otezla vs. Placebo und Etanercept vs. Placebo.
f Für PASI-50: p=0,0002 für Otezla vs. Placebo und p <0,0001 für Etanercept vs. Placebo.
g Für PASI-90: p=0,0169 für Otezla vs. Placebo und p=0,0009 für Etanercept vs. Placebo.
h Für sPGA-Score von befallsfrei oder nahezu befallsfrei: p=0,0005 für Otezla vs. Placebo und p <0,0001 für Etanercept vs. Placebo.
Morbus Behçet
Die Sicherheit und Wirksamkeit von Apremilast wurden in einer multizentrischen, randomisierten, placebokontrollierten Phase-3-Studie (RELIEF) an erwachsenen Patienten mit aktivem Morbus Behçet und oralen Ulcera untersucht. Die Patienten, die für eine systemische Therapie infrage kamen, hatten mindestens ein nicht-biologisches Morbus Behçet-Medikament gegen orale Ulcera erhalten: Colchicin (53%), NSAR (33%), andere Analgetika oder Anästhetika (18%), topische oder orale Kortikosteroide (14% bzw. 16%), Immunsuppressiva (14%). Die Patienten erfüllten die Kriterien der International Study Group (ISG) für Morbus Behçet. Die Patienten wiesen sowohl beim Screening als auch bei der Randomisierung mindestens zwei orale Ulcera auf. Patienten mit Morbus Behçet und aktiver Beteiligung wichtiger Organe (z.B. Manifestationen im Bereich der Augen, des kardiovaskulären Systems, des Gastrointestinaltrakts und des Zentralnervensystems), bei denen deshalb im Jahr vor dem Screening eine immunsuppressive Therapie erforderlich war, wurden aus der Studie ausgeschlossen. Eine Begleitbehandlung für Morbus Behçet war nicht erlaubt. Bei 37,7% der Patienten wurde eine begleitende Anwendung von Paracetamol oder NSAR angegeben.
Insgesamt wurden 207 Patienten mit Morbus Behçet im Verhältnis 1:1 auf eine Behandlung mit entweder Apremilast 30 mg zweimal täglich (n=104) oder Placebo (n=103) über 12 Wochen (placebokontrollierte Phase) randomisiert. Von Woche 12 bis 64 erhielten alle Patienten Apremilast 30 mg zweimal täglich (aktive Behandlungsphase).
Der primäre Endpunkt war die Fläche unter der Kurve (AUC) für die Anzahl der oralen Ulcera von der Baseline bis einschliesslich Woche 12. Die sekundären Endpunkte umfassten andere Parameter für orale Ulcera (Schmerzen durch orale Ulcera auf einer visuellen Analogskala (VAS)), Anteil von ulkusfreien Patienten (vollständiges Ansprechen), Zeit bis zum Beginn der Rückbildung oraler Ulcera und Anteil derPatienten, die eine Rückbildung von oralen Ulcera bis Woche 6 erreichten und über mindestens 6 weitere Wochen während der 12-wöchigen placebokontrollierten Behandlungsphase bei jedem Besuchstermin ulkusfrei blieben.
Der Altersbereich der Patienten reichte von 19 bis 72 Jahren, wobei das Durchschnittsalter bei 40 Jahren lag. Die durchschnittliche Dauer des Morbus Behçet betrug 6,84 Jahre. Alle Patienten hatten eine Vorgeschichte mit rezidivierenden oralen Ulcera, die aktuell aktiv waren. Ferner wiesen die Patienten in der Anamnese Hautläsionen (98,6%), genitale Ulcera (90,3%), muskuloskeletale Manifestationen (72,5%), Manifestationen im Bereich der Augen (17,4%), des Zentralnervensystems (9,7%) und des Gastrointestinatrakts (9,2%), Epididymitis (2,4%) und Gefässbeteiligung (1,4%) auf. Die mittlere Anzahl der oralen Ulcera bei Baseline lag bei 4,2 und 3,9 in der Apremilast- bzw. der Placebo-Gruppe.
Parameter der oralen Ulcera
Apremilast 30 mg zweimal täglich führte zu einer signifikanten Besserung oraler Ulcera, wie es durch die AUC für die Anzahl der oralen Ulcera von der Baseline bis einschliesslich Woche 12 (p<0,0001) im Vergleich zu Placebo gezeigt wurde.
In Woche 12 wurden signifikante Verbesserungen bei weiteren Parametern für orale Ulcera nachgewiesen.
Tabelle 7: Klinisches Ansprechen von oralen Ulcera in Woche 12 in der RELIEF-Studiea (ITT Population)
|
Endpunkt
|
Placebo
N=103
|
Apremilast
30 mg 2x tgl.
N=104
|
Absoluter angepasster Behandlungsunterschiedd
| |
AUCb für die Anzahl von oralen Ulcera von Baseline bis einschliesslich Woche 12 (ITT, MI)
|
LS-Mittelwert
222,14
|
LS-Mittelwert
129,54
|
92,60e
| |
Veränderung gegenüber Baseline bei den Schmerzen von oralen Ulcera gemessen anhand der VASc in Woche 12 (ITT, MMRM)
|
LS-Mittelwert
-18,7
|
LS-Mittelwert
-42,7
|
24,1e
| |
Anteil von Patienten mit Rückbildung von oralen Ulcera (ulkusfreier Mund) bis Woche 6, die über mindestens 6 weitere Wochen bei jedem Besuch während des 12-wöchigen placebokontrollierten Behandlungszeitraums ulkusfrei sind (ITT)
|
4,9%
|
29,8%
|
25,1%e
| |
Anteil der Patienten mit vollständigem Ansprechen der oralen Ulcera in der Woche 12 (ITT; NRI)
|
22,3%
|
52,9%
|
30,6%
|
a HR=Hazard Ratio; ITT=Intent-to-Treat; LS=kleinste Quadrate; MI=multiple Imputation; MMRM=Mischeffekte-Modell für wiederholte Messungen; NRI=Non-Responder Imputation.
b AUC=Fläche unter der Kurve.
c VAS=visuelle Analogskala; 0=keine Schmerzen; 100=die schlimmsten Schmerzen, die man sich vorstellen kann.
d Der angepasste Unterschied bei den Anteilen ist der gewichtete Durchschnitt der Behandlungsunterschiede über die 4 Strata der kombinierten Geschlechts- und Regionsfaktoren hinweg mit den Cochran-Mantel-Haenszel-Gewichtung.
e p-Wert <0,0001 für alle Apremilast vs. Placebo.
Von den 104 Patienten, die ursprünglich zu Apremilast 30 mg zweimal täglich randomisiert worden waren, behielten 75 Patienten (etwa 72%) diese Behandlung bis Woche 64 bei. Bei diesen Patienten wurde in der Behandlungsgruppe mit 30 mg Apremilast zweimal täglich bei jedem Besuch, bereits ab Woche 1, bis einschliesslich Woche 12, eine signifikante Reduktion der durchschnittlichen Anzahl von oralen Ulcera (p≤0,0015) sowie der Schmerzen durch orale Ulcera (p≤0,0035) im Vergleich zur Placebo-Gruppe beobachtet. Bei den Patienten, die durchgehend mit Apremilast behandelt wurden und in der Studie verblieben sind, blieb die Besserung der oralen Ulcera bis einschliesslich Woche 64 erhalten (Abbildung 1).
Bei den Patienten, die ursprünglich zu Apremilast 30 mg zweimal täglich randomisiert worden waren und in der Studie verblieben sind, blieb der Anteil der Patienten mit vollständigem Ansprechen der oralen Ulcera bis einschliesslich Woche 64 erhalten (53,3%).
Der klinische Nutzen von Apremilast 30 mg zweimal täglich wurde in verschiedenen Subgruppen nachgewiesen, die nach demographischen Ausgangsdaten, klinischen Ausgangsmerkmalen der Krankheit (einschliesslich Krankheitsdauer und Anzahl oraler Ulcera bei Baseline) und früherer Anwendung von Arzneimitteln zur Behandlung von Morbus Behçet definiert waren.
Abbildung 1: Mittlere Anzahl von oralen Ulcera nach Zeitpunkt bis einschliesslich Woche 64 (ITT-Population; DAO)
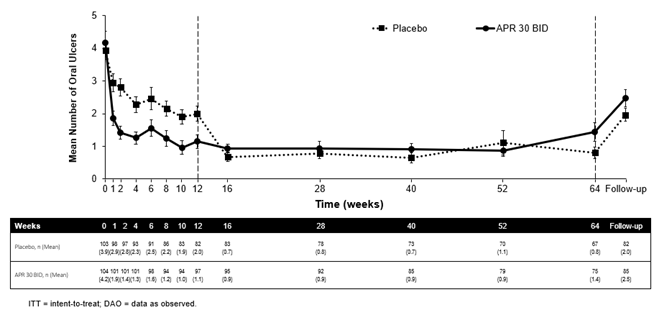
APR 30 BID=Apremilast 30 mg zweimal täglich;
Hinweis: Placebo bzw. APR 30 mg BID steht für die Behandlungsgruppe, in die die Patienten randomisiert wurden. Die Patienten der Placebo-Gruppe wurden in Woche 12 auf Apremilast 30 mg 2x tgl. umgestellt.
Der Follow-up-Zeitpunkt lag entweder 4 Wochen nach Abschluss der Studie in Woche 64 oder 4 Wochen nach vorzeitiger Beendigung der Behandlung vor Woche 64.
Gesamtverbesserungen der Krankheitsaktivität von Morbus Behçet
Apremilast 30 mg zweimal täglich führte zu einer signifikanten Abnahme der gesamthaften Krankheitsaktivität im Vergleich zu Placebo, wie die mittlere Veränderung gegenüber Baseline in Woche 12 beim Aktivitätsscore für das Behçet-Syndrom (BSAS) (p <0,0001), bei dem Formular für die aktuelle Morbus Behçet-Aktivität (BDCAF), dem aktuellen Morbus Behçet-Aktivitätsindex (BDCAI), der Wahrnehmung der Krankheitsaktivität durch den Patienten und der Gesamtwahrnehmung der Krankheitsaktivität durch den Kliniker gezeigt haben.
Bei den ursprünglich zu Apremilast 30 mg zweimal täglich randomisierten Patienten, die in der Studie verblieben sind, blieben die Verbesserungen (mittlere Veränderung gegenüber Baseline) beim BSAS und beim BDCAF bis Woche 64 erhalten.
Verbesserung der Lebensqualität
Apremilast 30 mg zweimal täglich führte im Vergleich zu Placebo zu einer signifikant grösseren Verbesserung der Lebensqualität (QoL), wie es in Woche 12 anhand des Fragebogens zur Lebensqualität bei Morbus Behçet nachgewiesen wurde.
Bei den ursprünglich zu Apremilast 30 mg zweimal täglich randomisierten Patienten, die in der Studie verblieben sind, blieb die Verbesserung der Lebensqualität bei Morbus Behçet bis Woche 64 erhalten.
PharmakokinetikAbsorption
Apremilast wird mit einer absoluten oralen Bioverfügbarkeit von ca. 73% gut resorbiert, wobei maximale Plasmakonzentrationen (Cmax) im Median nach ca. 2,5 Stunden (tmax) erreicht werden. Die Pharmakokinetik von Apremilast ist linear und zeigt einen dosisproportionalen Anstieg der systemischen Exposition im Dosisbereich von 10 bis 100 mg täglich. Die Akkumulation von Apremilast ist bei einmal täglicher Gabe minimal und beträgt bei zweimal täglicher Gabe bei gesunden Probanden ca. 53% und bei Psoriasis-Patienten 68%. Gleichzeitige Nahrungsaufnahme verändert die Bioverfügbarkeit nicht. Daher kann Apremilast zu den Mahlzeiten oder unabhängig davon eingenommen werden.
Distribution
Die Plasmaproteinbindung von Apremilast beträgt beim Menschen ca. 68%. Das mittlere scheinbare Verteilungsvolumen (Vd) beträgt 87 Liter, was auf eine extravaskuläre Verteilung hindeutet.
Metabolismus
Apremilast wird sowohl über CYP-vermittelte als auch über nicht-CYP-vermittelte Stoffwechselwege umfassend metabolisiert, und zwar unter anderem durch Oxidation, Hydrolyse und Konjugation. Dies lässt darauf schliessen, dass es bei Hemmung eines einzelnen Eliminationsweges wahrscheinlich zu keiner ausgeprägten Arzneimittelinteraktion kommt. Der oxidative Metabolismus von Apremilast wird primär von CYP3A4 getragen, wobei auch CYP1A2 und CYP2A6 geringfügige Beiträge leisten. Nach oraler Verabreichung ist Apremilast die Hauptkomponente im Blutkreislauf. Apremilast wird umfassend metabolisiert: Lediglich 3% resp. 4% der verabreichten Muttersubstanz werden im Urin bzw. den Fäzes wiedergefunden. Der zirkulierende Hauptmetabolit ist das Glucuronidkonjugat von O-demethyliertem Apremilast (M12, inaktiv).
In vitro ist Apremilast weder ein Inhibitor noch ein Induktor von Cytochrom-P450-Enzymen. Daher ist es unwahrscheinlich, dass Apremilast bei gleichzeitiger Verabreichung mit Substraten von CYP-Enzymen die Clearance von oder Exposition gegenüber Arzneistoffen beeinflusst, welche durch CYP-Enzyme metabolisiert werden.
In vitro ist Apremilast ein Substrat und ein schwacher Inhibitor von P-Glycoprotein (IC >50 μM).
In vitro besitzt Apremilast eine nur geringfügige oder keine Hemmwirkung (IC50 >10 μM) auf die organischen Anionentransporter (OAT) 1 und 3, den organischen Kationentransporter (OCT) 2, die organischen Anionen-Transport-Polypeptide (OATP) 1B1 und 1B3 oder das BCRP (Breast Cancer Resistance Protein) und ist kein Substrat für diese Transporter. Daher sind klinisch relevante Arzneimittelinteraktionen unwahrscheinlich, wenn Apremilast zusammen mit Arzneistoffen verabreicht wird, die Substrate oder Inhibitoren dieser Transporter sind.
Elimination
Die Plasma-Clearance von Apremilast beträgt bei gesunden Probanden im Durchschnitt etwa 10 l/h, mit einer terminalen Eliminationshalbwertszeit von ca. 9 Stunden. Nach oraler Verabreichung von radioaktiv markiertem Apremilast werden etwa 58% resp. 39% der Radioaktivität im Urin bzw. in den Fäzes wiedergefunden, wobei etwa 3% resp. 4% der radioaktiven Dosis als Apremilast im Urin bzw. in den Fäzes wiedergefunden werden.
Kinetik spezieller Patientengruppen
Pharmakokinetik bei älteren Patienten
Apremilast wurde bei jungen und älteren gesunden Probanden untersucht. Die Apremilast-Exposition bei älteren Probanden (65 bis 85 Jahre) ist bei der AUC etwa 13% und bei der Cmax etwa 6% höher als bei jungen Probanden (18 bis 55 Jahre). Eine Dosierungsanpassung ist bei älteren Patienten nicht erforderlich. Es gibt nur wenige pharmakokinetische Daten für Patienten im Alter von mehr als 75 Jahren.
Pharmakokinetik bei Niereninsuffizienz
Zwischen Personen mit leichter oder mässiger Einschränkung der Nierenfunktion und entsprechenden nierengesunden Probanden (jeweils N=8) besteht bei der Pharmakokinetik von Apremilast kein bedeutsamer Unterschied.
Bei Patienten mit leichter Einschränkung der Nierenfunktion verkleinerte sich die AUC von Apremilast um ~14%, während sich die Cmax um ~6% erhöhte; bei Patienten mit mittelschwerer Einschränkung der Nierenfunktion vergrösserte sich die AUC um ~23%, während die Cmax um ~13% abnahm.
Bei 8 Probanden mit schwerer Niereninsuffizienz, welchen eine Einzeldosis 30 mg Apremilast verabreicht wurde, erhöhte sich die AUC resp. Cmax von Apremilast um ca. 89% bzw. 42%. Bei Patienten mit schwerer Niereninsuffizienz (eGFR unter 30 ml/min/1,73 m2 oder CLcr <30 ml/min) ist die Apremilast-Dosierung auf 30 mg einmal täglich zu reduzieren. Bei Dialysepatienten wurden keine Studien durchgeführt.
Pharmakokinetik bei Leberinsuffizienz
Die Pharmakokinetik von Apremilast und seinem Hauptmetaboliten M12 wird durch eine mässige oder schwere Einschränkung der Leberfunktion nicht beeinflusst. Bei Patienten mit Leberinsuffizienz ist keine Dosierungsanpassung erforderlich.
Präklinische DatenFertilität und frühembryonale Entwicklung
In einer Fertilitätsstudie an männlichen Mäusen hatte Apremilast in oralen Dosierungen von bis zu 50 mg/kg/Tag (entsprechend ca. dem 3-Fachen der klinischen Exposition) keine Auswirkungen auf die männliche Fertilität.
In einer Studie, in der mit oralen Dosierungen von 10, 20, 40 und 80 mg/kg/Tag die Toxizität in Bezug auf die Fertilität weiblicher Mäuse und die embryofötale Entwicklung untersucht wurden, wurden ab einer Dosierung von 20 mg/kg/Tag eine Verlängerung der Östruszyklen eine längere Zeit bis zur Paarung und Postimplantationsverluste beobachtet. Dennoch paarten sich alle Mäuse und die Trächtigkeitsraten waren unbeeinflusst. Die NOEL (No Observed Effect Level)-Dosis für die weibliche Fertilität lag bei 10 mg/kg/Tag (dem 1,0-Fachen der klinischen Exposition).
Embryofötale Entwicklung
In einer Studie, in der mit oralen Dosierungen von 10, 20, 40 und 80 mg/kg/Tag die Toxizität in Bezug auf die Fertilität weiblicher Mäuse und die embryofötale Entwicklung untersucht wurden, waren die absoluten und/oder relativen Herzgewichte der Muttertiere bei Dosierungen von ≥20 mg/kg/Tag erhöht. Vermehrte Frühresorptionen und Ossifikationsverzögerung wurden bei ≥20 mg/kg/Tag beobachtet. Verminderte Fötengewichte und eine verzögerte Ossifikation des Os supraoccipitale des Schädels wurden bei ≥40 mg/kg/Tag beobachtet. Die NOEL-Dosis für Muttertiere und die embryofötale Entwicklung betrug bei der Maus 10 mg/kg/Tag (das 1,3-Fache der klinischen Exposition). Bis zur höchsten Dosierung von 80 mg/kg/Tag (dem 4,0-Fachen der klinischen Exposition) wurden keine behandlungsbedingten Entwicklungsmissbildungen beobachtet.
In einer bei Affen mit oralen Dosierungen von 20, 50, 200 und 1000 mg/kg/Tag durchgeführten Studie zur embryofötalen Entwicklungstoxizität führten Dosierungen ab 50 mg/kg/Tag zu einem dosisabhängigen Anstieg pränataler Verluste (Aborte). Kein auf das Prüfpräparat zurückzuführender Effekt im Hinblick auf pränatale Verluste wurde bei 20 mg/kg/Tag (dem 1,4-Fachen der klinischen Exposition) beobachtet. Bis zur höchsten in dieser Studie geprüften Dosierung von 1000 mg/kg/Tag (dem 3,5-Fachen der klinischen Exposition) wurden beim Affen keine behandlungsbedingten Auswirkungen auf die fötale Entwicklung oder Missbildungen beobachtet. Die abortierten Feten wurden jedoch nicht untersucht.
Prä- und postnatale Entwicklung
In einer Prä- und Postnatalstudie wurde Apremilast trächtigen Mäusen in Dosierungen von 10, 80 und 300 mg/kg/Tag vom 6. Trächtigkeitstag bis zum 20. Tag der Laktationsperiode oral verabreicht. Abnahmen des Körpergewichts und verminderte Gewichtszunahme bei den Muttertieren sowie ein tödlicher Verlauf im Zusammenhang mit Wurfkomplikationen wurden bei 300 mg/kg/Tag beobachtet. Klinische Zeichen einer maternalen Toxizität im Zusammenhang mit dem Werfen der Jungen wurden auch bei jeweils einer Maus unter 80 und 300 mg/kg/Tag beobachtet. Eine erhöhte peri- und postnatale Mortalität und verminderte Körpergewichte der Jungtiere in der ersten Woche der Laktation wurden bei Dosierungen ≥80 mg/kg/Tag (dem ≥4,0-Fachen der klinischen Exposition) beobachtet. Es fanden sich keine auf Apremilast zurückzuführenden Wirkungen auf die Trächtigkeitsdauer, die Anzahl trächtiger Mäuse am Ende der Gestationsperiode und die Anzahl der Mäuse mit einem Wurf sowie keine Auswirkungen auf die Entwicklung der Jungtiere nach dem 7. postnatalen Tag. Die Mortalität und die Auswirkungen auf die Entwicklung der Jungtiere, die in der ersten Woche der Postnatalperiode beobachtet wurden, standen wahrscheinlich im Zusammenhang mit der auf Apremilast zurückzuführenden Toxizität für die Jungtiere (vermindertes Gewicht und verminderte Überlebensfähigkeit der Jungen) und/oder mit einer mangelnden Versorgung der Jungen durch das Muttertier (erhöhte Inzidenz eines fehlenden Nachweises von Milch im Magen der Jungtiere). Alle Auswirkungen auf die Entwicklung wurden in der ersten Woche der Postnatalperiode beobachtet. Keine weiteren auf Apremilast zurückzuführenden Effekte wurden in den weiteren Entwicklungsphasen vor und nach der Entwöhnung beobachtet, einschliesslich der Parameter der sexuellen Reifung, des allgemeinen Verhaltens und des Paarungsverhaltens, der Fertilität und des Uterus. Die NOEL-Dosis betrug bei der Maus für die maternale Toxizität und die F1-Generation 10 mg/kg/Tag (das 1,3-Fache der klinischen Exposition [AUC]).
Karzinogenität
Karzinogenitätsstudien bei Mäusen (bei Dosierungen bis zu 1000 mg/kg/Tag entsprechend dem 8,8-Fachen der klinischen Exposition) und Ratten (bis zu 20 mg/kg/Tag in männlichen Mäusen entsprechend dem 0,08-Fachen der klinischen Exposition und bis zu 3 mg/kg/Tag in weiblichen Tieren entsprechend dem 1,1-Fachen der klinischen Exposition) ergaben keinen Anhalt für eine auf die Behandlung mit Apremilast zurückzuführende Karzinogenität.
Genotoxizität
Apremilast ist nicht genotoxisch.
Sonstige HinweiseHaltbarkeit
Das Arzneimittel darf nur bis zu dem auf der Packung mit «EXP» bezeichneten Datum verwendet werden.
Besondere Lagerungshinweise
In der Originalverpackung, nicht über 30 °C und ausser Reichweite von Kindern aufbewahren.
Zulassungsnummer65346 (Swissmedic).
PackungenOtezla Startpackung (4x 10 mg, 4x 20 mg, 19x 30 mg) mit insgesamt 27 Filmtabletten [B]
Otezla 30 mg: Packungen mit 56 Filmtabletten [B]
ZulassungsinhaberinAmgen Switzerland AG, Risch
Domizil: 6343 Rotkreuz
Stand der InformationAugust 2020.
Version#290420
|